Apart from deciding whether the length of the prediction is similar to the majority of the lengths of the BLAST hits, there are many other validation tests that have to be applied on the predicted data. This week we have been focusing on reading frame inconsistency, detecting gene merges and duplications.
- Gene duplications - can occur due to an error in the replication mechanism of the cell, or simply due to a sequencing error, which we have to report. In these situations BLAST finds more high-scoring segment pairs (hsp) from a single hit. Here's an example for PB16966 gene (of Yannick's knowledge), where a sequence of 6 exons is repeated. BLAST found the following:
<Hit_num>1</Hit_num>
<Hsp_num>1</Hsp_num>
<Hsp_query-from>1715</Hsp_query-from>
<Hsp_query-to>3346</Hsp_query-to>
<Hsp_hit-from>6</Hsp_hit-from>
<Hsp_hit-to>1642</Hsp_hit-to>
<Hsp_num>2</Hsp_num>
<Hsp_query-from>10</Hsp_query-from>
<Hsp_query-to>1630</Hsp_query-to>
<Hsp_hit-from>1</Hsp_hit-from>
<Hsp_hit-to>1642</Hsp_hit-to>
Notice that in such cases hsps from the same hit do overlap and the matched regions in the predictions do not. - Reading frame inconsistency - this case can be retrieved from the BLAST output: for a sequence of nucleotides, we use blastx to find the most similar proteins. The query sequence is translated in six reading frames, and all the
resulting six-protein sequences are compared (there's no evidence where the codon starts from, so all the 3 possibilities of starting the gene translation are considered in BLAST; also another 3 possibilities when reading the sequence in reverse). There can be a problem in the predicted sequence when the hits have various reading frames, of the same sign (sequences are read in the same direction). The sequencing errors leading to this type of errors are usually reading frame shifts. For example a
nucleotide was accidentally introduced at a certain point of the mRNA
extraction, causing a reading frame inconsistency between the left and
the right part pf the predicted gene, but still read in the same direction.
- When reading frame is consistent for all the hits, we consider 3 cases:
- One direction gene merge (----> ----------->) - same sign reading frames for all hits
- Opposite direction gene merge (----><----------) - two opposite sign reading frames for the hits
- otherwise we have no merge
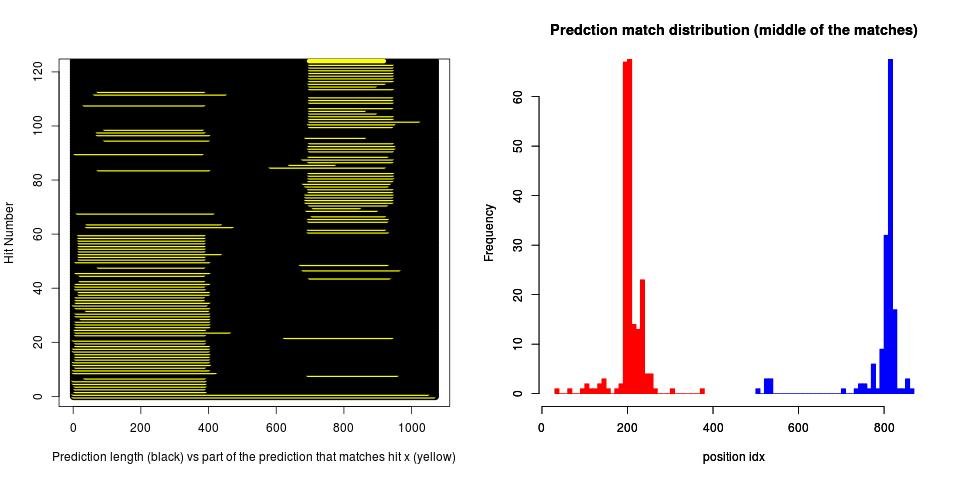
Our approach for identifying gene merges is to analyze the distribution of the middles of each region in the predicted gene that matches a hit gene from BLAST. You can observe in the plots below that in case of gene merge the data have a multimodal distribution, otherwise the distribution is unimodal. We calculate a score that quantifies whether the assumption of unimodality is more appropriate than the one for multimodality using the approach described in [3] for model based clustering. More plots here [2].
1) Example for merged genes (bimodal distribution)
2) No merge (unimodal distribution)

0 comments:
Post a Comment